






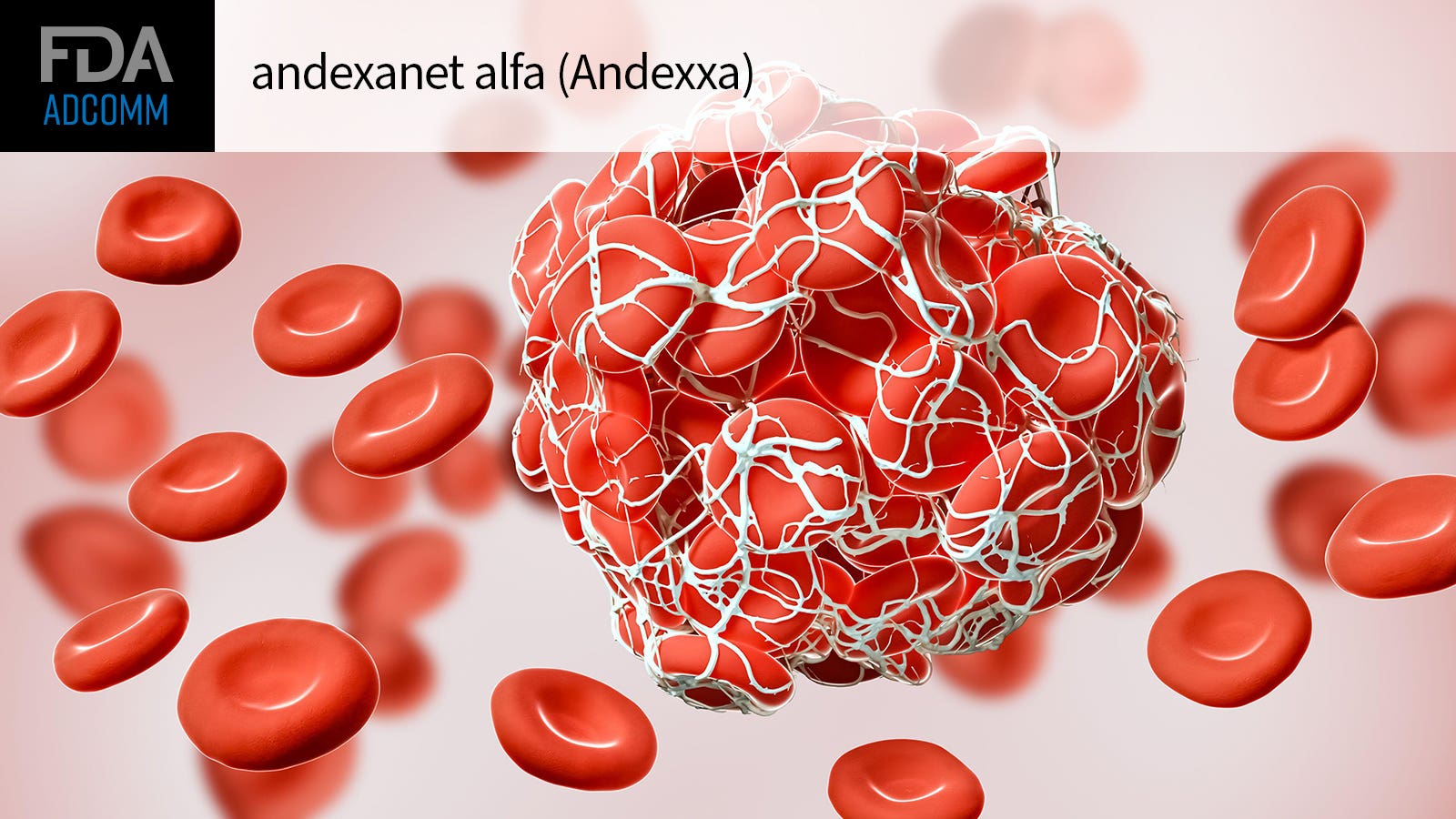
FDA advisors pushed for nuance in discussing whether andexanet alfa (Andexxa) should gain full approval, citing uncertainties about which dose and in which patients it could be best used to stop bleeding.
Members of the Cellular, Tissue, and Gene Therapies Advisory Committee agreed on Thursday that andexanet has a demonstrated ability to control hematoma volume change at 12 hours based on the ANNEXA-I trial of direct oral anticoagulant (DOAC) users with intracerebral hemorrhage (ICH).
But they were less sure if that is truly meaningful, without other supporting evidence of clinical benefit.
With this year’s ANNEXA-I trial, manufacturer AstraZeneca was able to fulfill the FDA’s request for a randomized controlled trial of andexanet with a clinical endpoint. The trial showed that the drug helped people with acute ICH on factor Xa (FXa) inhibitors curb expansion of hematoma volume. This appeared to be andexanet’s main benefit, as it was no help in improving neurologic status or survival.
“It’s a real regulatory dilemma here,” said panelist Donald Kohn, MD, of the University of California Los Angeles. “They met their primary endpoint but the primary endpoint wasn’t designed to look at how a patient feels, functions, or survives … The MRI findings of volume is just really a surrogate marker.”
Joseph Wu, MD, PhD, of Stanford University in California, noted that “obviously the drug works, but we’re confronted by the lack of clinical data … and maybe also an increase in thrombosis. And that’s what we’re really stuck at.”
Safety factored heavily in Thursday’s FDA advisory committee meeting discussion, as ANNEXA-I also tied the drug to thrombotic risks. By day 30 in the trial, 14.6% of participants receiving andexanet logged thrombotic events, compared with 6.9% in the usual care group typically receiving a prothrombin complex concentrate (PCC). Thrombosis-related deaths occurred in 2.5% versus 0.9%, respectively.
Notably, as early as 2016’s ANNEXA-4 initial study presentation, outside observers had noted a signal of excess thrombotic events among andexanet users.
Even so, the interim results of ANNEXA-4 were enough for andexanet to win FDA accelerated approval in 2018 on the basis of the surrogate endpoint of change in anti-activated FXa activity in the study.
“There is no alternative here,” stressed committee member Sanjay Ahuja, MD, MSc, MBA, of Innovative Hematology in Indianapolis. “People use PCCs for reversal of these agents without any approval …. Physicians will use PCCs the way they want to use PCCs, and I think that might be indirectly contributing to harm. Once you have an approved drug in this category, I think it’s only beneficial for the patient population in general. It makes the lives of physicians easier. It makes insurance approvals easier.”
Andexanet still holds the indication of reversal of anticoagulation due to life-threatening or uncontrolled bleeding among patients treated with rivaroxaban (Xarelto) or apixaban (Eliquis). The advisory panel members were not asked to vote on further regulatory action for andexanet.
“I’m wondering if the indications can be more nuanced,” said Evan Snyder, MD, PhD, of Sanford Burnham Prebys Medical Discovery Institute in La Jolla, California.
“Maybe when a clinician has this drug … they would know that a patient at high risk for a thrombotic event should not be given this drug, whereas those who are on DOACs for another indication would be the ones that would benefit from this drug. We can be more nuanced with the indication of this drug once it’s in the toolbox, like we do with so many drugs,” Snyder suggested.
Andexanet is a recombinant modified human FXa protein that binds FXa inhibitors to restore thrombin generation.
In ANNEXA-I, patients randomized to the andexanet arm were assigned to either a low-dose or high-dose regimen, based on the timing and dosage of their last DOAC dose. Hemostatic efficacy, the primary endpoint, was a composite of expansion of hematoma by 35% or less, an increase of less than 7 points on the National Institutes of Health Stroke Scale, and no receipt of rescue therapy.
While the FDA is not required to follow the advice of its advisory committees, it typically does.
![author['full_name']](https://clf1.medpagetoday.com/media/images/author/nicoleLou_188.jpg)








+ There are no comments
Add yours